Mechanics
The simulation is an agent-based 2D spatio-temporal simulation. Cells are agents. Each cell has a string, the genome, which unambigously encodes a set or proteins, the proteome. These proteins can change molecules in and around the cell. Through that each cell can process information. Cell proteomes can form complex networks with hubs, feedback loops, cascades, and oscillators. When randomly changing a cell genomes, their networks change randomly, too. By selectively replicating certain cells while killing others, cells can evolve.
- Genetics explains how genetics work in this simulation
- Chemistry explanation for molecules, reactions and energy coupling
- Kinetics explains the reaction kinetics in this simulation
I believe that most interesting behaviors take many time steps to evolve. That's why this simulation is implemented with millions of time steps in mind. Genetics, chemistry, physics in this simulation is simplified a lot. It is a tradeoff between a reasonable amount of complexity and performance.
Genetics
All mechanisms are similar to bacterial translation. A cell's genome is a chain of nucleotides represented by a string of letters T, C, G, A. There are start and stop codons which define coding regions (CDSs). Transcription can start at any start codon and will end when the first in-frame stop codon encountered. CDSs without a stop codon are not considered [James NR 2016]. This is different to real-life transcription in that there are no promotor and terminator sequences. Each CDS is translated into one protein, giving each cell a certain proteome. What each protein can do is defined by its domains.
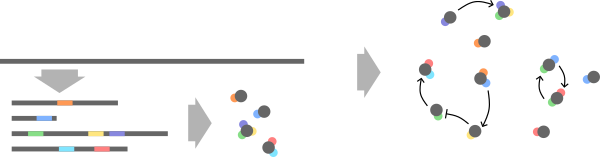
A cells genome is transcribed into multiple coding regions, each of which forms a protein. Each coding region can contain multiple domains of which each has a different purpose and different kinetics. Together these proteins form signal processing networks in the cell.
Currently, there are three domain types: catalytic, transporter, regulatory. Each domain consists of a region of genetic code that defines the domain type itself and several regions that define its further specifications. What these specifications are depends on the domain type.
Catalytic domains can catalyze one reaction each. All possible reactions are defined in the simulation's chemistry (see Chemistry). Specifications are the catalyzed reaction, affinities for substrates and products, maximum velocity, domain orientation (see Energy).
Transporter domains can move a molecule species across the cell membrane, i.e. from the outside world into the cell and vice versa. All possible molecule species are defined in the simulation's chemistry (see Chemistry). Specifications are the molecule species, molecule affinity, maximum velocity, domain orientation (see Energy).
Regulatory domains can regulate a protein through an effector molecule. A protein with only a regulatory domain has no function. But if the protein also has a catalytic or transporter domain, the regulatory domain can up- or down-regulate this domain. Specifications are the effector molecule species, a hill coefficient, whether it is activating or inhibiting, the affinity of the effector.
Affinity and maximum velocity are kinetic parameters which are described in Kinetics in more detail. Which specific combination of nucleotides encodes which specific parameter, e.g. a particular maximum velocity value, is defined by mappings. These mapping are created when the Genetics and Kinetics objects are initialized. A user can also override them later on.
The rules of this genetic setup are quite simple. E.g. there is no transcriptional and post-transcriptional regulation. Abundance, location, and efficiency of CDSs are not taken into consideration. Yet still this simple genetic setup can already create complex protein networks with hubs, feedback loops, and cascades. Some example transcriptomes and biochemical patterns are shown in figures 2 and figures 7.
Chemistry
As the basis of this simulation one has to define which molecule species exist and which reactions are possible. Molecule species are defined with attributes about how fast they can diffuse and permeate, and with an internal energy value. This energy is the hypothetical energy that this molecule would release if it was fully deconstructed. Reactions define how molecule species can be converted. They are all reversible (see Kinetics for details). Here, the simulation takes Gibbs free energy as an analogy:
where is the standard Gibbs free energy of the reaction, is the energy of substrate with stoichiometric coefficient , and is the energy of product with stoichiometric coefficient . It is used to calculate an equilibrium constant for this reaction.
where is the gas constant and is the absolute temperature. As further described in Kinetics the reaction quotient always moves towards . So, reactions tend to be favourable into the direction which deconstructs high energy molecules and constructs low energy molecules.
However, all reactions catalyzed by the same protein are considered to be energetically coupled. All domains are considered when calculating . Thus, an energetically unfavourable reaction can progress if it is powered by an energetically favourable one. In which orientation these domains are energetically coupled is defined in the domain itself as a region that encodes a boolean . All domains with orientation work from left to right, and vice versa for . E.g. if there are 2 catalytic domains and , they would become if they have the same orientation, and if not.
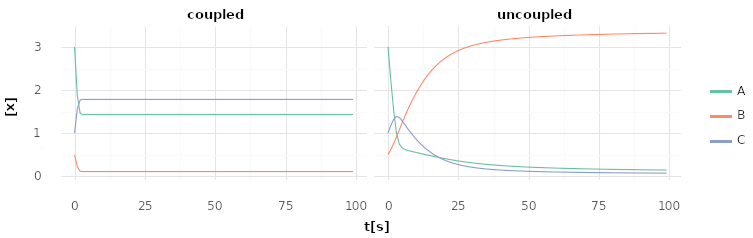
Uncoupled and coupled catalysis over time. Molecule concentrations inside a cell while it catalyzes 3A C | -10 kJ and A + B C | 5 kJ each step. Left shows a cell with a single protein with 2 domains which are energetically coupled as 4A + B 2C | -5 kJ. Right shows a cell with 2 proteins with a single domain each for both reactions respectively.
Transporter domains are also involved this way. A transporter is seen as a reaction that converts an intracellular molecule to its extracellular version (and vice versa). For a transporter is always zero only drives the reaction. Thus, a transporter can drive a reaction while molecules are allowed to diffuse along their concentration gradient, or a reaction might drive a transporter to pump molecules against their concentration gratient.
Kinetics
All reactions in this simulation are based on Michaelis-Menten-Kinetics. However, the possibility of a reverse reaction is allowed on the second step. In the simplest case enzyme (E) forms a complex (ES) with substrate (S) and modifies it to product (P). P can bind E again and react back to S.
Where , , , are the forward and reverse rates for the first and second step. This can be described with the reversible Michaelis-Menten equation
with
where velocity is the change in over time, is the concentration of total enzyme ( and ), is the substrate concentration, is the product concentration.
In the simulation S and P can be molecules, and the enzyme is a protein with some functional domains. For simplification is assumed to be constant for all cells and proteins, and . The reversible Michaelis-Menten equation can then be re-written as
defines the maximum velocity of the protein. and describe reciprocal affinities to S and P. In general a protein can consist of catalytic, transporter, and regulatory domains (see Genetics). Transporters are treated as catalytic domains which convert a molecule species from its intracellular version to its extracellular one and vice versa. Regulatory domains regulate the protein non-competitively. The final velocity of a protein is
with
where is allosteric regulation (details below), is the concentration of substrate with stoichiometric coefficient , and is the concentration of product with stoichiometric coefficient . Concentration change over time of any molecule species can be calculated by multiplying its stoichiometric number (using IUPAC nomenclatur) with . Over time the reaction will approach an equilibrium state where , and its reaction quotient (the equilibirum constant):
Thus, defines in which direction the reaction will proceed. As described in Chemistry is calculated from an analogy of the reaction's Gibbs free energy. Actual values for , , are derived from the domain specifications (see Genetics). One part of of the domain specification encodes maximum velocity . Another part encodes affinity from which and are derived.
Allosteric regulation with effector molecules A is modeled as
where is the concentration of effector molecule , is the hill coefficient, and is the effector concentration producing half occupation. for activating effectors, for inhibiting effectors. Proteins without regulatory domains are always allowed to be active. Some kinetics examples are given in figures 6.
When values for the mappings of nucleotide sequences to values for maximum velocities and affinities
are created, they are sampled from distributions with user defined boundaries.
For multiple domains specifications with overlapping
values for , , are averaged.
The resulting distributions of these values are shown in
figures 11.
All defaults were chosen with some assumptions in mind:
(1) molecule numbers (such as in world.molecule_map) represent mM,
(2) a time step represents 1 second,
(3) molecule energies represent Joules.